












































【大师兄带你读文献】之如何通过读文献提升科研思维和课题设计能力?选题思路实战篇6

 WOrange 推荐
WOrange 推荐大师兄带你读文献学习课题设计系列
读文献学课题设计系列前帖分享内容传送门:
今天给大家解析一篇Cell Metabolism大子刊上的胞葬作用通过代谢物调控巨噬细胞介导免疫反应研究论文,抽丝剥茧看清该研究是如何进行课题研究方向切入及work model逻辑串联的。
此次我们将结合课题逻辑简要模式箭头示意图,更清晰把握文献逻辑脉络构建及证明。
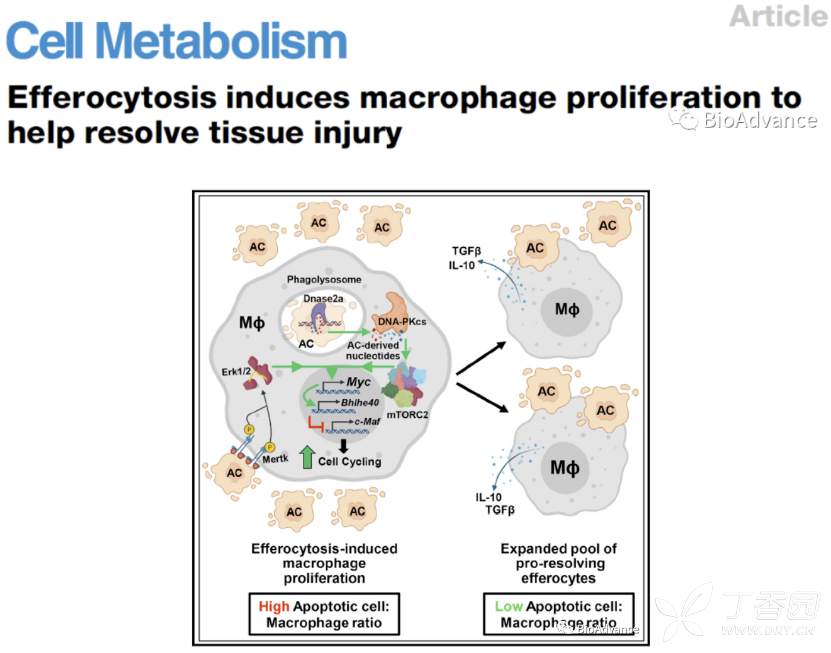
本研究证实的work model如下
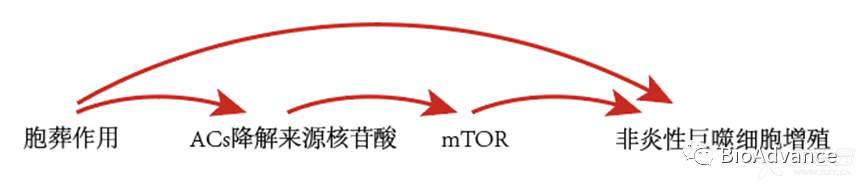
胞葬作用-ACs降解核苷酸-PKcs-mTORC2/Rictor-Myc-巨噬细胞增殖
下面通过问题导向,解析调控模式如何实现
1. 为何选择胞葬作用对巨噬细胞增殖作为课题切入点?
我们知道细胞死亡包括程序性死亡(如凋亡)和坏死两种类型,其中,凋亡过程中细胞萎缩形成凋亡小体被巨噬细胞清除,细胞内物质不会释放到组织间隙诱发炎症反应;而坏死则不同,会导致细胞破裂,释放大量胞内物质,诱发炎症反应,导致组织损伤。胞葬作用主要是指,巨噬细胞吞噬凋亡的细胞,阻止细胞坏死发生,因而可以阻断炎症反应,保护组织免受炎性损伤,促进炎性损伤组织修复。
该课题组长期致力于研究胞葬作用在组织炎性损伤修复中的作用(该课题组关于胞葬作用研究已发论文如下图所示)。








通过前期十多年间的研究积累,已经明确证实胞葬作用可以诱导巨噬细胞的免疫抑制表型(分泌TGFβ、IL-10)促进组织炎性损伤修复。
免疫细胞调控生物学过程主要通过两种方式:
- 细胞功能表型改变,如M1和M2表型极化;
- 细胞数量改变,如增殖扩增。
该组前期研究中已经发现凋亡细胞(Apoptosis cells, ACs)被巨噬细胞吞噬发生胞葬作用后,ACs在巨噬细胞中被溶酶体水解释放的氨基酸、脂质等物质可以导致巨噬细胞转变为免疫抑制表型,从而促进炎性组织修复,这方面的研究已经非常多。而关于胞葬作用对巨噬细胞增殖的调控研究很少,潜在机制不明。考虑到胞葬作用促进巨噬细胞介导的炎性组织损伤修复,除了通过已报道的诱导巨噬细胞免疫抑制表型,逻辑上应该还存在胞葬作用通过某种机制促进免疫抑制性巨噬细胞增殖,以增强炎性损伤修复,因此确立了胞葬作用可能促进免疫抑制性巨噬细胞增殖这一科学假设,此科学假设便是本研究最关键的创新性表型切入点。
2. 如何确定胞葬作用促进巨噬细胞增殖(efferocytosis-induced macrophage proliferation, EPIM)这一关键表型?
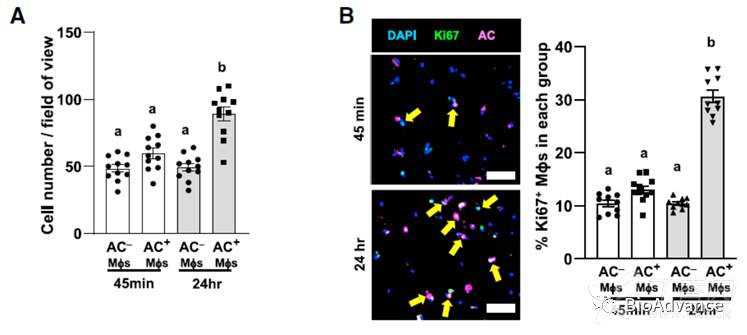
将巨噬细胞(本研究利用BMDM)与凋亡中的细胞(本研究选择的是Jurkat细胞系)共培养,之后利用Ki67、Edu标记增殖细胞等方式证实胞葬作用会促进增强巨噬细胞增殖。由此得到了本研究最核心创新性表型,即胞葬作用可以促进巨噬细胞增殖如下模式所示:

3. 如何证实胞葬作用通过凋亡细胞在胞葬作用中降解后产生的核苷酸促进巨噬细胞增殖?
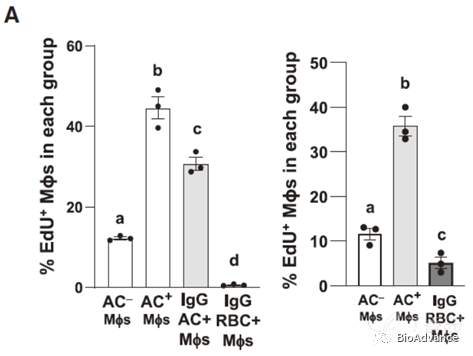
该课题组及其他团队之前研究已经发现胞葬作用中,ACs降解产生的氨基酸和脂类都可以促进巨噬细胞免疫抑制表型。我们知道细胞降解产物中除了氨基酸、脂类,还包含大量的核苷酸物质,那么ACs降解来源的核苷酸是否在胞葬作用促巨噬细胞增殖中发挥关键作用呢。为了证实这一假设,本研究做了三个关键实验:1.将lgG包被的红血细胞(RBC)与巨噬细胞进行共培养(红细胞无细胞核,胞葬作用降解后无核苷酸),结果显示并不能诱导EIMP。2. DNase2a是参与将吞噬酶体中的DNA降解为核苷酸。siRNA沉默DNase2a导致AC+巨噬细胞EdU掺入减少,细胞增殖减少,SCF1则没有明显变化(阻断细胞降解中DNA降解为核苷酸)。3.将外源性的核苷酸电转入RBC中,共培养后则可以诱导EIMP(外源性导入核苷酸)。以上结论证实胞葬作用确实可以通过ACs降解来源的核苷酸促进巨噬细胞增殖,如下模式所示:

4. 如何证实胞葬作用通过降解来源的核苷酸激活mTORC2/Rictor促进巨噬细胞增殖?
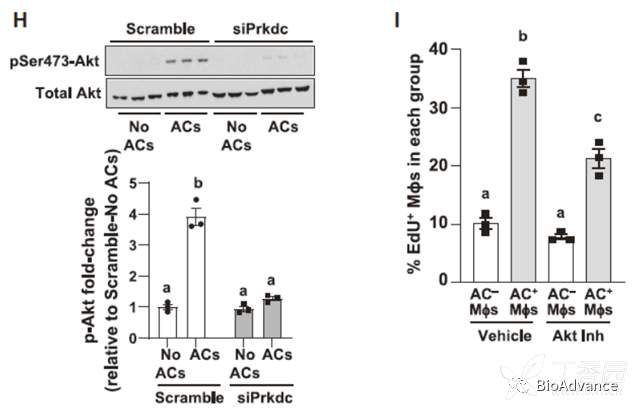
接着问题是,胞葬作用通过ACs降解产生的核苷酸如何促进巨噬细胞增殖的?此处基于文献报道可知DNA依赖的蛋白激酶(dna - pk)既可以结合寡聚核苷酸,又可以激活mTOR进而促进细胞增殖。而胞葬作用中ACs降解产生的就是寡聚核苷酸,因此提出科学假设:胞葬作用中ACs降解的核苷酸很可能通过与dna-pk结合,进而激活mTOR(激活后p-AKT增加),促进巨噬细胞增殖,并通过体内、体外敲降和抑制剂实验证实了以上假设。
逻辑上完成了以下模式:
5. 如何证实胞葬作用通过降解来源的核苷酸激活mTORC2/Rictor后再通过Myc促进巨噬细胞增殖?
基于该课题组之前已发表文章发现,胞葬作用诱导巨噬增殖是通过激活Myc表达介导,因此,想到mTOR激活后应该也很可能通过Myc这一关键转录因子促进巨噬细胞增殖。
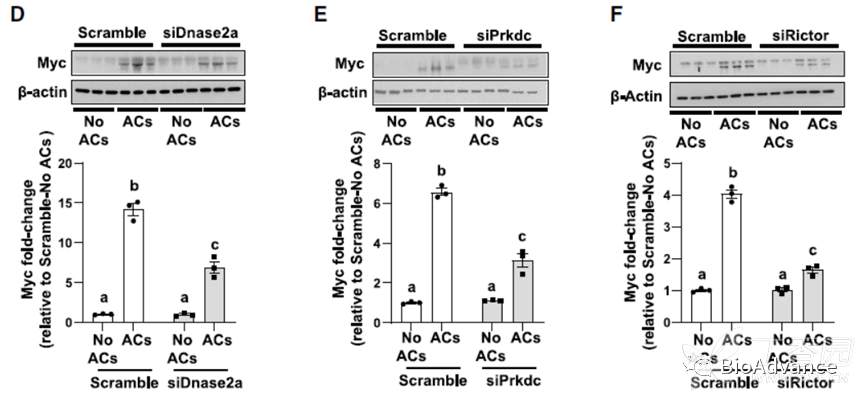
接着也通过一系列实验证实胞葬作用降解来源核苷酸激活的mTOR信号确实可以激活Myc高表达,并通过rescue实验证实Myc介导了mTOR的促巨噬细胞增殖作用。
逻辑上完成了一下模式:
以上就是整篇文章课题选择及逻辑串联思路解析。
--------------------------------------------------分割线---------------------------------------------------------
时间允许的话,可以继续阅读下文中关于该文献的详细内容解读。
背景及摘要
巨噬细胞在组织炎性损伤修复过程中发挥着重要作用。组织炎症损伤修复的一个关键过程就是巨噬细胞对凋亡细胞(ACs)的清除,这个过程又称作胞葬作用。ACs激活巨噬细胞中受体介导的信号通路,启动巨噬细胞胞葬作用,防止凋亡细胞坏死来阻止组织炎症,并促进组织损伤修复。相反,在许多慢性炎症疾病(包括进展性动脉粥样硬化)中,胞葬作用会出现缺陷,导致组织损伤和慢性炎症。巨噬细胞通过胞葬作用促炎性损伤修复功能受细胞内特定代谢途径的影响,其中胞葬期间ACs在巨噬细胞中被吞噬溶酶体降解产生的代谢产物(如氨基酸和脂质)发挥重要作用,然而胞葬作用产生的哪种代谢物是如何激活巨噬细胞的促炎性损伤修复的机制尚未充分阐明。
本研究发现ACs胞葬期间,ACs的DNA被巨噬细胞吞噬溶酶体中的DNase2a降解为核苷酸,核苷酸通过DNA-PKcs-mTORC2/Rictor轴增加Myc表达,进而促进非炎性巨噬细胞增殖,发挥抑制炎性环境促进损伤修复作用。
本文亮点
- 巨噬细胞吞噬凋亡细胞后增殖能力增强;
- 凋亡细胞衍生的核苷酸激活巨噬细胞的DNA-PKmTorc2-Myc信号轴;
- 胞葬巨噬细胞的增殖促进了炎症的消散;
- 阻断巨噬细胞的增殖可阻碍动脉粥样硬化的消退。
研究结果
1、胞葬的巨噬细胞会发生增殖
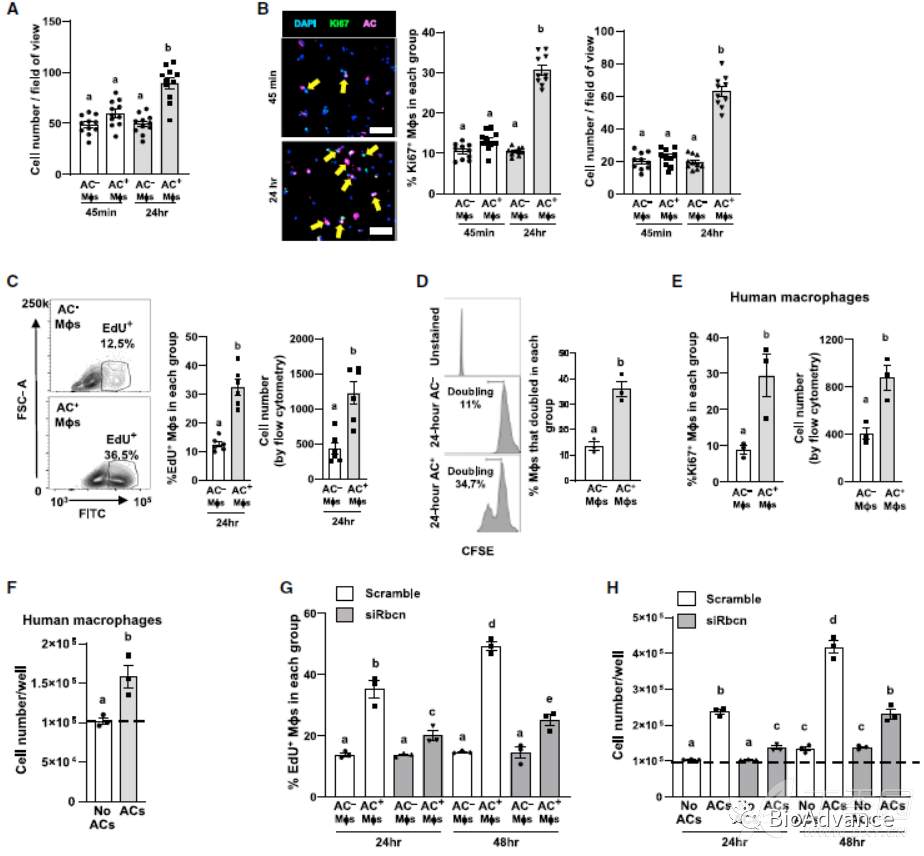
为了探究胞葬巨噬细胞与普通巨噬细胞的差异,作者将骨髓来源的巨噬细胞(BMDM)与Vybrant did标记的凋亡Jurkat细胞共培养45min。结果显示,含有AC的巨噬细胞增多,而不含AC的巨噬细胞没有明显变化。含有AC的巨噬细胞,表达增殖标志物Ki67+水平增加。EDU及CFSE实验也得出一致的结果。作者在人来源的骨髓巨噬细胞中也观察到了一致的现象(HMDM)。类似的,IL-4处理也能促进巨噬细胞增殖,但LPS及IFN-γ不能诱导巨噬细胞增殖。综上所述,胞葬可以触发小鼠和人巨噬细胞的增殖,但不会触发炎性巨噬细胞的增殖。这个过程我们称之为胞葬诱导的巨噬细胞增殖(EIMP)。
2、胞葬巨噬细胞的增殖需要ACs的吞噬酶体降解,并有MerTK的额外刺激
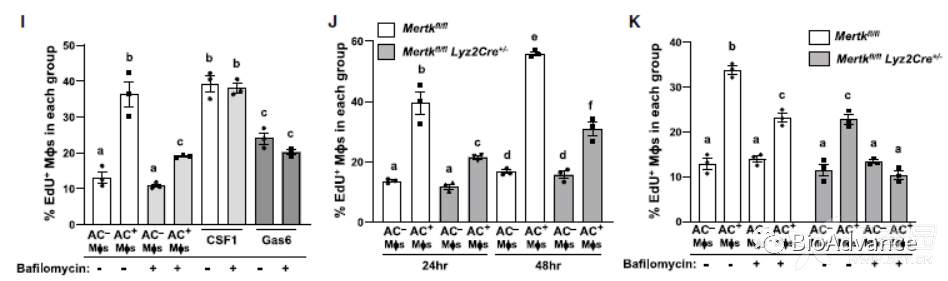
为了探究胞葬过程中吞噬酶体的作用,作者进行了Rubicon(一种对吞噬酶体功能非常重要的LC3吞噬作用相关的支架蛋白)的干扰实验(siRbcn)。结果显示,尽管siRbcn后对AC的摄取没有明显的影响,但胞葬巨噬细胞的增殖受到显著抑制。接下来,作者又进行了Bafilomycin(巴菲霉素,一种可以吞噬酶体的酸化、AC内化降解的抑制剂)的实验,一致的,也阻断了AC+巨噬细胞中edu阳性的百分比。相反,CSF1作为一个重要的对照,其诱导的巨噬细胞增殖不涉及吞噬酶体,也不能被Bafilomycin阻断。
作者观察到,虽然阻断ACs吞噬酶体降解显著抑制EIMP,但仍有残留的AC诱导的增殖反应(Fig 1I)。为了解释这一发现,作者假设EIMP可能与AC对细胞信号受体的初始激活有关,因此作者聚焦到了胞葬的受体MerTK。在缺乏MerTK的巨噬细胞中,ACs诱导的巨噬细胞增殖存在defect,而CSF1诱导的巨噬细胞增殖则没有明显的变化。此外,巴菲霉素治疗和MerTK缺失的联合治疗与单独两种方法相比,显著消除了AC+巨噬细胞中edu -阳性的百分比增加。因此,EIMP存在两条独立的途径,1是通过吞噬酶体降解内化AC;2是通过MerTK受体途径。
3、EIMP需要吞噬酶体释放AC来源的寡核苷酸,寡核苷酸通过DNA-PKcs激活mTORC2
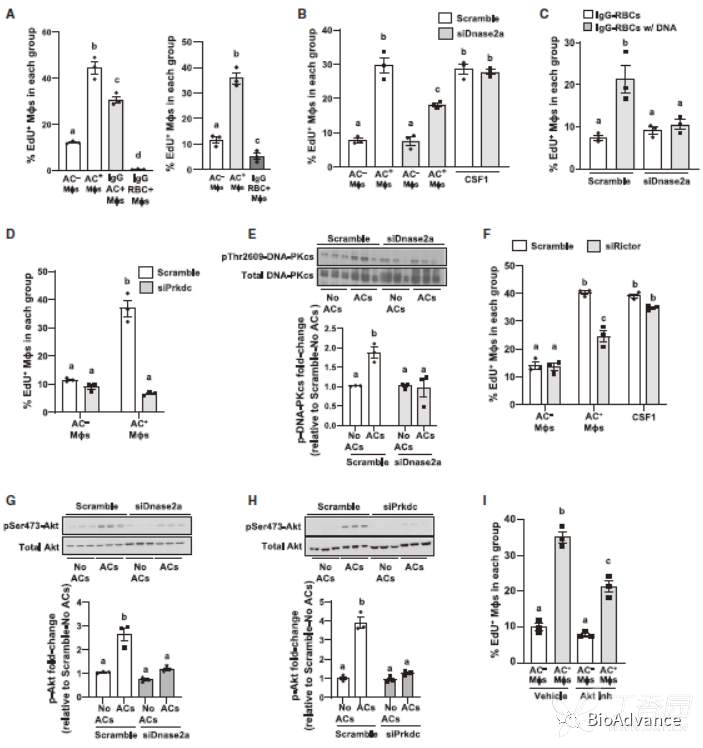
作者前述结果发现,EIMP的一个重要的表型是细胞周期的活化,即增殖。作者假设可能是由于AC来源的核苷酸触发了EIMP。因此,作者设计了以下实验。1.将lgG包被的红血细胞(RBC)与巨噬细胞进行共培养,结果显示并不能诱导EIMP。2. DNase2a是参与将吞噬酶体中的DNA降解为核苷酸;siRNA沉默DNase2a导致AC+巨噬细胞EdU掺入减少,细胞增殖减少,SCF1则没有明显变化。3.将外源性的核酸电转入RBC中,共培养后则可以诱导EIMP。综上所述,这些结果表明,吞噬酶体DNase2a产生的ac衍生寡核苷酸对EIMP是必要的。
为了揭示Dnase2a衍生的寡核苷酸和EIMP之间的机制联系,我们寻找了能够结合DNA寡核苷酸并与增殖相关的酶,最终锁定DNA依赖的蛋白激酶(dna - pk)。DNA-PK,它由一个催化亚基DNA-PKcs(基因名Prkdc)和一个核苷酸结合域组成。已有研究表明,Prkdc可以通过激活mTORC促进细胞增殖。与猜想一致,沉默Prkdc后,巨噬细胞EIMP减少;且沉默DNase2a后dna – pk的蛋白水平也会减少。接下来,作者进一步探索了dna - pkcs介导的AC核苷酸在EIMP中的信号转导作用。沉默Rictor(mTORC中的6个成员之一,控制其激酶活性),对AC的摄取没有影响,但导致EIMP显著减少。此外,ACs诱导Akt在丝氨酸-473位点磷酸化,这是mTORC诱导增殖必需的信号,使用泛Akt抑制剂MK-2206抑制Akt,同样可以抑制EIMP。而siDNase2a and siPrkdc则可以阻断其诱导的EIMP。以上结果证实了以下通路在EIMP中的作用:dnase2a水解AC-DNA/DNA-PKcs / mTORC2 / phospho-Akt /proliferation。
4、增加mTORC2和MerTK-ERK1/2信号下游的Myc表达对EIMP是必要的
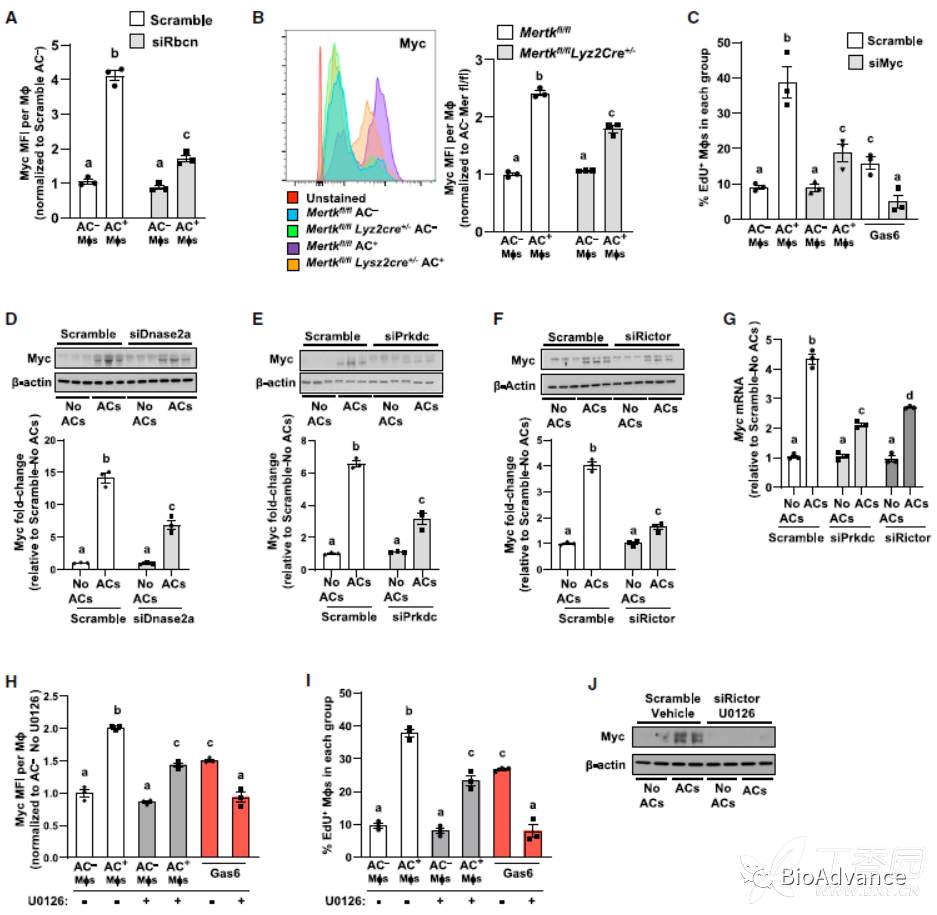
作者前期研究发现MerTK和吞噬酶体-核苷酸途径都集中于促增殖转录因子Myc促进EIMP。作者也发现,含有AC的巨噬细胞的myc水平显著高于不含AC的巨噬细胞,而沉默Rbnc和MerTK后,myc水平减少。进一步的,沉默myc后,AC+巨噬细胞的增殖明显受到抑制。因此,ACs的吞噬酶体降解和MerTK的参与都会增加Myc的表达以刺激EIMP。
关于吞噬酶体途径,作者继续探究DNase2a-DNA-PKcs-mTORC2途径是否可能导致Myc增加。事实上,DNase2a、DNA-PKcs或Rictor的沉默导致Myc蛋白和Myc mRNA的降低。
关于MerTK,作者假设ERK1/2的激活可能参与其中,因为目前研究发现MerTK参与导致巨噬细胞的ERK1/2的激活。与预期一致,AC+巨噬细胞磷酸化erk1 /2增加,MerTK敲除则可以rescue。利用ERK磷酸化抑制剂U0126处理巨噬细胞,显示ERK抑制剂抑制Myc表达。
综上,吞噬酶体和MerTK通路分别通过mTORC2和ERK1/2共同增加胞葬巨噬细胞中的Myc,进而促进EIMP。
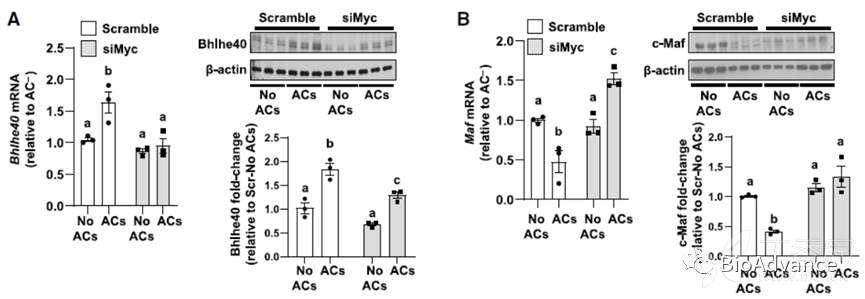
5、Myc通过上调Bhlhe40和下调c-Maf来驱动EIMP
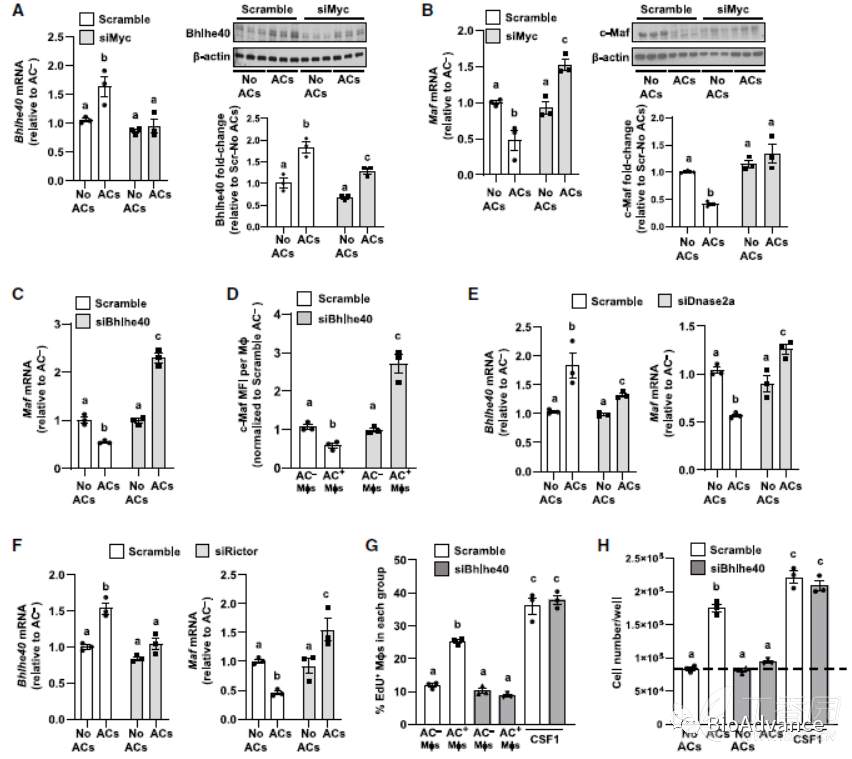
文献研究表明,在巨噬细胞增殖的不同环境中,Myc靶向Bhlhe40下调抗增殖蛋白c-Maf水平,促进细胞增殖。因此,作者探究了Myc是否可能通过增加Bhlhe40的表达来驱动EIMP。含有AC的巨噬细胞Bhlhe40的转录及蛋白水平显著增加,沉默myc后,则显著减少。此外,含有AC的巨噬细胞Maf mRNA和c-Maf蛋白降低,而Myc沉默可阻止这些降低。此外,无论是DNase2a还是Rictor沉默都阻止了AC诱导的Bhlhe40增加和c-Maf降低。最重要的是,沉默Bhlhe40完全阻止了AC+巨噬细胞的增殖,而csf1诱导的增殖未见此现象。综上所述,AC介导的MerTK-ERK信号激活以及DNase2a衍生的AC寡核苷酸激活DNAPKcs- mTORC2信号,诱导Myc高表达Bhlhe40,Bhlhe40抑制细c-Maf,促进细胞增殖。
6、EIMP的巨噬细胞在体外是TGFb和IL-10的生产者和效应者
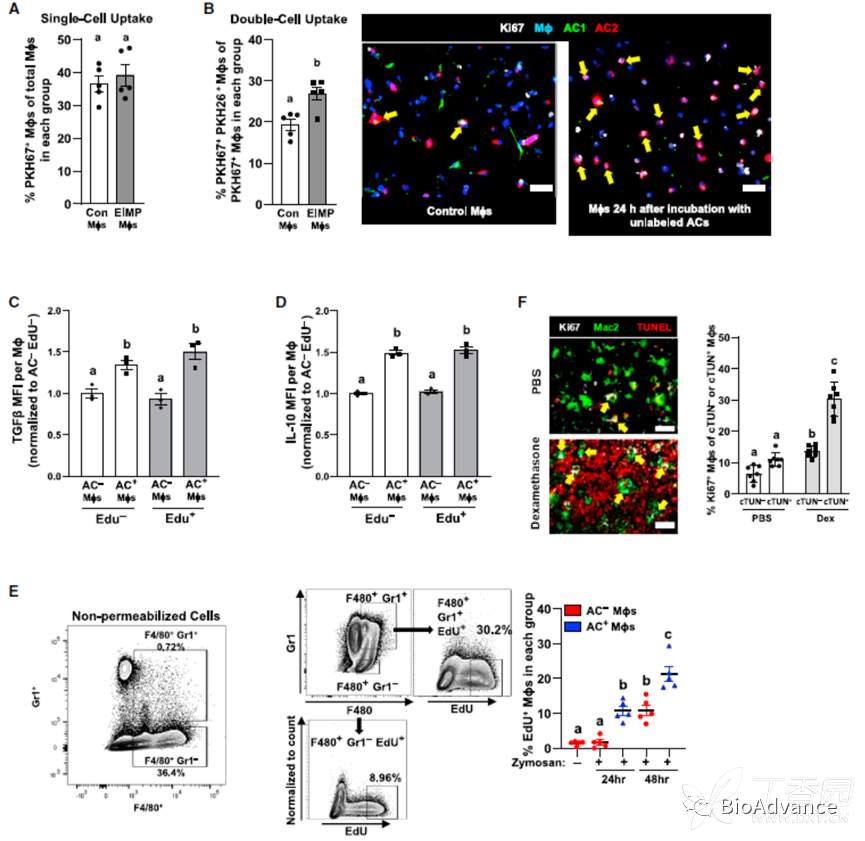
搞清楚EIMP的分子机制后,接下来作者继续探究了机体的EIMP会导致怎样的结局呢?作者猜测,EIMP可以扩大巨噬细胞池,促进巨噬细胞吞噬AC的能力,并产生更多的效应分子。一致的,作者通过标记AC实验,发现EIMP后巨噬细胞吞噬AC能力变强。且TGF-β及IL-10的水平升高。
7、体内模型中验证EIMP现象及其对组织的修复作用
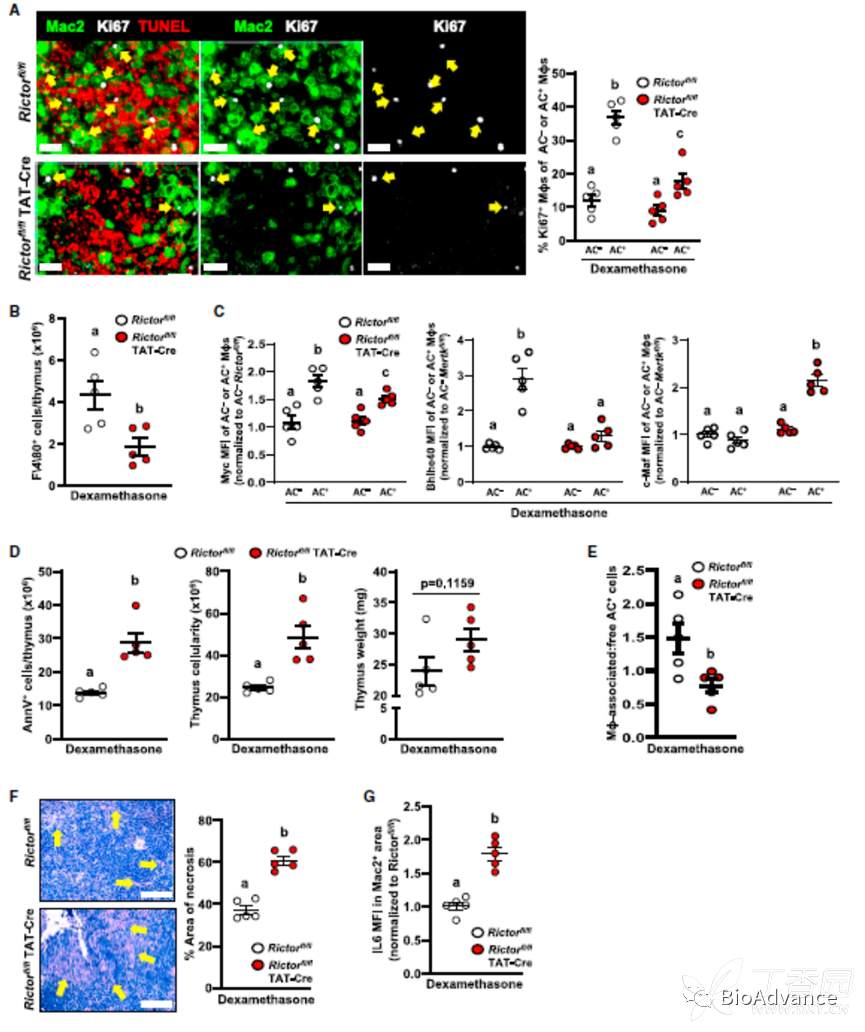
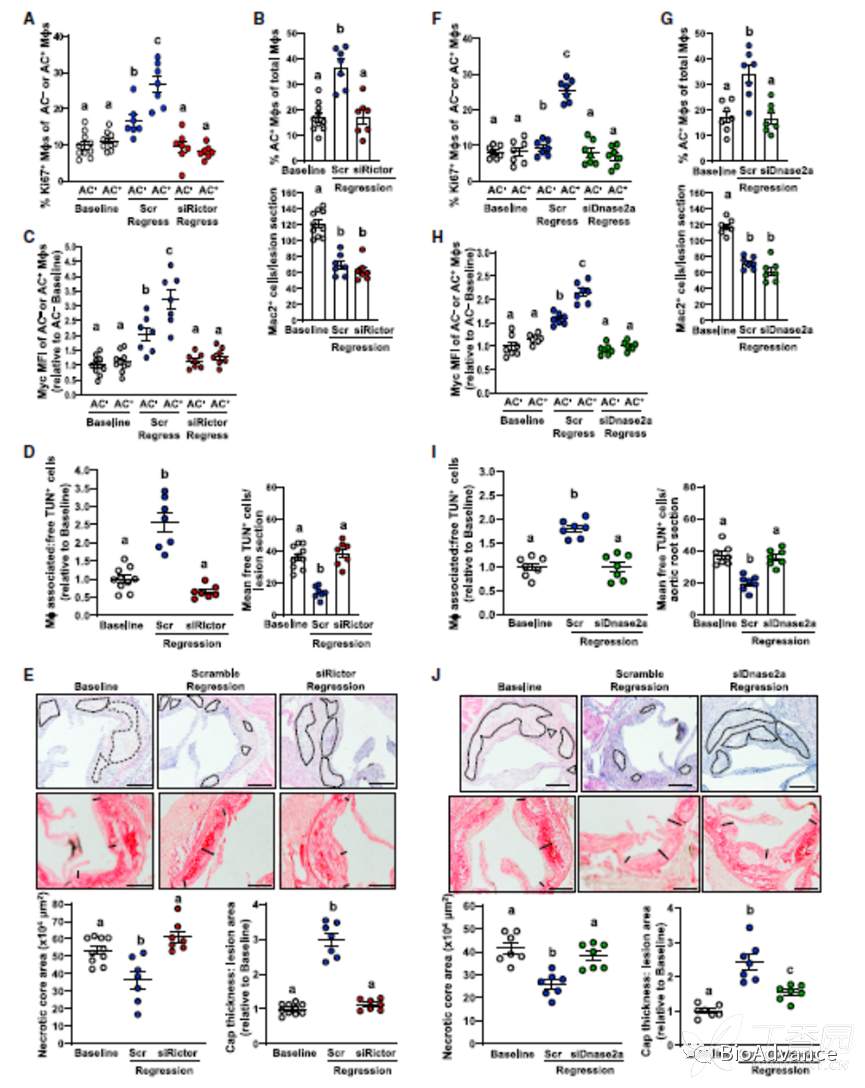
前面的实验都是在体外得到的结果,接下来,作者在体内对前面的结果进行了验证。作者一共用了3种模型进行验证:1酵母菌素A诱导的急性腹膜炎模型;2 地塞米松诱导的胸腺细胞凋亡模型;3 动脉粥样硬化模型。
急性腹膜炎模型中,炎症前期会诱导中性粒细胞(Neu)浸润,对菌体进行杀伤;然后中性粒细胞发生凋亡,此时,巨噬细胞会吞噬凋亡的中性粒细胞。作者在造模前对小鼠进行EDU,以标记增殖的细胞。造模后,利用F4/80+GR1-标记未吞噬neu的巨噬细胞,F4/80+GR1+标记发生了胞葬的巨噬细胞。结果显示,发生胞葬的巨噬细胞增殖水平显著升高。
在地塞米松诱导的胸腺细胞凋亡模型中。作者验证了myc-Bhlhe40-Maf信号通路。并发现EIMP在体内具有清除AC和保护组织的作用。敲除Rictor的巨噬细胞则会导致胸腺炎症及病理情况加剧。
在动脉粥样硬化模型中,作者利用纳米颗粒包裹的siRNA靶向干预体内巨噬细胞的特定分子水平(DNaes2a,Rictor),发现破坏EIMP的形成,会导致组织坏死区域及炎症水平减少。
全文总结
本研究发现,胞葬作用可以促进非炎性巨噬细胞增殖,增强免疫抑制表型,提出了并证实了一个work model:凋亡细胞(ACs)被巨噬细胞吞噬胞葬期间,ACs的DNA被巨噬细胞吞噬溶酶体中的DNase2a降解为核苷酸,核苷酸通过DNA-PKcs-mTORC2/Rictor轴增加Myc表达,Myc通过上调Bhlhe40和下调c-Maf来驱动EIMP(胞葬诱导的非炎性巨噬细胞增殖),进而促进非炎性巨噬细胞增殖,形成免疫抑制(胞葬作用-ACs降解核苷酸-PKcs-mTORC2/Rictor-Myc-巨噬细胞增殖)。纵观全文,最大的创新点是发现了胞葬过程可以促进非炎性巨噬细胞增殖促进炎性损伤修复表型。并通过扎实的体外、体内实验揭示了潜在分子机制。
以上就是本次文献思路解析及精读的全部内容,如有所收获记得点赞、收藏、关注哟,同时也可以关注大师兄个人专栏(科研心路历程、科研课题设计思路、SCI写作投稿)!
对文献精读解析感兴趣的小伙伴,下次见咯~


