












































【转录组测序】序列比对及组装

1,概要
使用指定的基因组作为参考进行序列比对及后续分析
人类参考基因组下载地址见:ftp://ftp.ensembl.org/pub/release-80/fasta/homo_sapiens/
HISAT2 是一个来自 RNA 测序实验 reads 的高效比对系统,TopHat2 / Bowtie2 的继任者。HISAT 使用一个基于 Burrows-Wheeler 变换和 Ferragina-Manzini(FM)索引的索引方案,利用了两类索引进行比对:一个全基因组FM索引以定位每个比对,和许多局部 FM 索引用于非常快地扩展这些比对,实现了更快的速度和更少的资源占用。
利用 StringTie 对比对上的 reads 进行组装,StringTie 是基于最优化理论建立的算法,利用比对信息构建多可变剪切图谱运用构建流量网络从而根据最大流量算法来对 reads 进行组装和评估其表达量,同 Cufflinks 等其他软件相比可以构建更完整的转录本和更好的评估表达量。
基本流程:
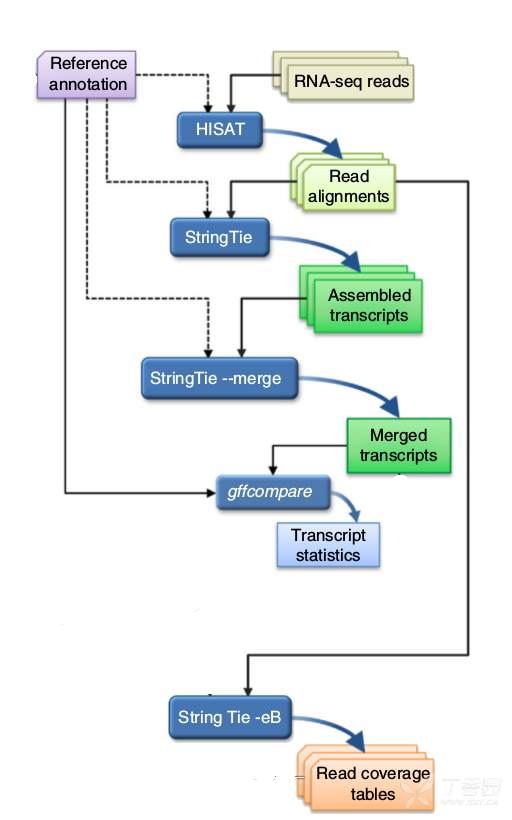
基本流程:
1.1 下载
#1.下载 http://www.ccb.jhu.edu/software/hisat/downloads/
#选择一个版本即可,这里选择2.0.4二进制版本
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.0.4-Linux_x86_64.zip
#解压直接使用
主要使用hisat2 和 hisat2-build工具
1.2 建索引
hisat2-2.0.4/hisat2-build -p 4 Homo_sapiens.GRCh38.genome.fa Homo_sapiens.GRCh38.genome
1.3 hisat2比对
hisat/2.0.4/hisat2 --dta -p 6 --rna-strandness RF -x Homo_sapiens.GRCh38.genome -1 Human_I694-01-L11_good_1.fq -2 Human_I694-01-L11_good_2.fq -S Hisat/L11/L11.HISAT_aln.sam > Hisat/L11/L11.log
1.4 sam转bam
samtools/0.1.18/samtools view -F 4 -Su Hisat/L11/L11.HISAT_aln.sam | samtools/0.1.18/samtools sort - Hisat/L11/L11.HISAT_aln.sorted
1.5 bam建index
samtools/0.1.18/samtools index L11.HISAT_aln.sorted.bam L11.HISAT_aln.sorted.bam.bai
1.6 stringtie进行序列组装
stringtie Hisat/L11/L11.HISAT_aln.sorted.bam -G Homo_sapiens.GRCh38.gff3 -p 6 -l L11 -o StringTie/L01/StringTie_asm.gtf


