












































引发长期发热的Still病的诊断与治疗

前言
不明原因发热是一个非常具有挑战的临床难题。而Still病(SD)是这类不明原因发热疾病谱里的重要疾病种类。
在过去,该病分别命名为全身型幼年特发性关节炎(sJIA)和成人期Still病(AOSD);2024年,欧洲抗风湿病联盟(EULAR)/欧洲儿科风湿病学会(PReS)联合发布指南,建议把其共同命名为Still病[1]。
在《长期发热的Still病有哪些临床特征,需要如何识别?》介绍了SD的临床表现,本期文章讨论诊断与治疗。
什么时候怀疑Still病?
参考2024年发布的EULAR/PReS联合指南,符合如下情况者应怀疑SD[1]:
- 一日1次或一日2次的高峰热(≥39°C),持续≥7天;
- 伴随发热而出现的皮疹;
- 有可能出现关节痛/肌痛;但不是必然的;
- 高炎症提示:中性粒细胞白细胞增多、红细胞沉降率(ESR)升高、血清C反应蛋白(CRP)和铁蛋白偏高。
然而,和上述表现相似的疾病有很多。感染性疾病、肿瘤、炎症性疾病等等都可能。医生首先要注意的点是,SD患者会发生自发性的体温下降,表现为大多数患者在没有药物干预下体温下降至正常,小部分会降低到低热(<38.3℃)[2]。
自发性的体温下降——这是SD与很多发热疾病截然不同的点。持续高热,体温不干预便不下降,这可否定SD。
注意,小部分患者在刚开始不是这样的发病模式,而是持续发热,或者完全不发热,但最终会走向这样有自发体温下降的高峰热模式[2]。
在刚发病时,SD患者还可有咽痛[3-4],这点成人SD患者更常见到[4]。而SD患者后续反复发作时,咽痛发生率就显著下降[5]。
在过去,常常用铁蛋白显著增高来提示SD,但是的确有小部分SD患者的铁蛋白仅仅是稍增高,幅度不大[6]。再考虑到很多发热疾病都有铁蛋白增高,该检测的特异性不够。
因此,在今天一旦临床怀疑(特定发热模式、白细胞增高、CRP高等)就应该立即检测血清白细胞介素(IL)18、S100 A8/A9(也称为血清钙卫蛋白)和S100 A12[1]。
为与其他疾病鉴别,医生应仔细完善病原学检测、淋巴结活检等,这有利于和感染性疾病、血液肿瘤等疾病鉴别。
注意并发MAS
SD患者可以在病程中的任何时间点,突发致命性并发症:巨噬细胞活化综合征(MAS)[1]。这个时间点可能是:刚SD诊断时、SD病情得到良好控制时、SD病情控制好后的减药过程中……
而动态检测铁蛋白数值的变化,有助于早期发现MAS。对于疑似MAS,或MAS高风险的SD患者,应每天,甚至一天内两次检测铁蛋白[1]。
血清sIL-2R、CXCL-9、IL-18水平、腺苷脱氨酶2(ADA2)活性、活化的CD8 T淋巴细胞百分比,或可能活化的CD4+或单核细胞,是新的评估MAS的血清检测项目,它们对诊断MAS具有较高的敏感性、特异性[1,7]。
对于MAS的临床表现、诊断与治疗,将专题讨论。此处不再赘述。
鉴别诊断
任何的诊断都是一个不断的反复的鉴别诊断过程。在诊断SD时,医生要仔细鉴别那些可能的混淆疾病。
1
腺苷脱氨酶2缺乏症
腺苷脱氨酶2缺乏症(DADA2)是一个可以引起发热、血管炎、淋巴结肿大等问题的自身炎症性疾病。
DADA2的发热没有固定模式,但可能一段时间里和SD相似,因此该病有可能与SD混淆[8]。我国天津市儿童医院风湿科曾报道一例DADA2与儿童SD混淆的病例[9]。
DADA2患者的ADA2活性严重下降,几乎是0或者接近于0[8],而SD患者并发MAS时,ADA2是增高的[1,7]。另外,川崎病患者的ADA2也是正常的[7]。
(补充阅读:罕见病挑战(8):DADA2的诊断与治疗)
2
其他单基因自身炎症性疾病
与DADA2类似,很多自身炎症性疾病都存着长程发热,比如,家族性地中海热(FMF)、高IgD综合征(也叫MKD)、肿瘤坏死因子(TNF)受体1相关周期性综合征(TRAPS)、冷炎素周期性发热综合征(CAPS)、VEXAS综合征等等。
但是,多数情况下,这些疾病的发热模式与SD颇为不同。虽然可能少数病人会在某个段时间内发生与SD相似的发热模式。对上述疾病颇为熟悉的情况下,与SD鉴别并不困难。
而且,新近研究证实,IL-18水平可以协助与这些疾病鉴别[10]。
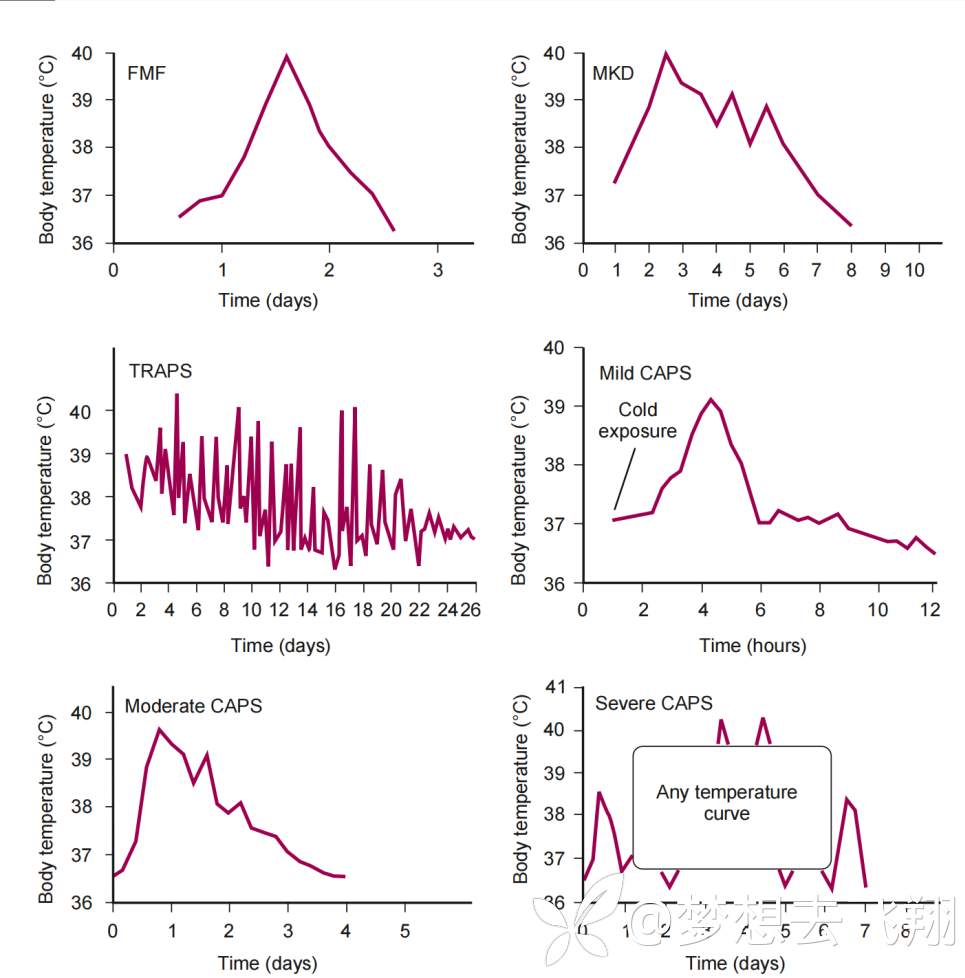
图1. 经典的自身炎症性疾病的发热模式[11]
3
LACC1缺陷相关幼年特发性关节炎
幼年特发性关节炎(JIA)通常被认为是一个多基因疾病。然而,2015年一项研究发现了LACC1缺陷相关的全身型幼年特发性关节炎(sJIA)——儿童发病的SD[12]。
LACC1也叫FAMIN,是一种在髓系细胞中表达的47-kD蛋白[13-14]。
尽管已经有多个LACC1缺陷相关幼年特发性关节炎的报道[15-18],比如,武汉儿童医院就曾于2023年[17]、深圳市儿童医院于2024年都有报道[18],但是研究未能发现LACC1与炎症小体、干扰素等关联起来,而是注意到它和巨噬细胞有一定关联[19]。其具体是如何引发JIA,还待进一步研究。
目前尚无确定LACC1缺陷相关幼年特发性关节炎的临床特征,比如,武汉儿童医院、深圳市儿童医院报道的病例都没有显著的发热,但在其他国家的报道里,却有很多类似SD发病的JIA病例。笔者建议,对于任何JIA,包括幼年起病的SD都应怀疑该病可能。
4
自身免疫性淋巴细胞增生性综合征
自身免疫性淋巴细胞增生性综合征(ALPS)通常在出生后头几年内发病,最常见的首发临床表现是非感染性非恶性淋巴细胞增生(表现为淋巴结肿大、脾肿大和/或肝肿大)和自身免疫性血细胞减少(包括血小板减少和溶血性贫血)。淋巴瘤是其晚发并发症[20]。该病也有相对突出的IL-18增高[20],这点与SD相似。儿童SD务必考虑到和ALPS鉴别。
5
Castleman病
Castleman病也叫血管滤泡性淋巴结增生,是一组具有共同组织病理学特征的异质性淋巴细胞增生性疾病。该病大体可以分3个类型。临床上主要表现为发热、淋巴结肿大等。该病有相对突出的IL-6增高[2]。但该病的IL-18也可能增高,但似乎没有SD突出。对Castleman病熟悉的医生,不难与SD鉴别。
6
血管内淋巴瘤
血管内淋巴瘤(IVL)是一种极其罕见的非霍奇金淋巴瘤亚型,因为该病是小血管腔内淋巴瘤细胞增生,特别是在毛细血管和毛细血管后微静脉内,血管外无明显肿瘤团块,外周血也检测不到循环肿瘤细胞[2]。所以该病诊断相当具有挑战性。
IVL的临床表现多种多样,主要以发热、体重减轻、乏力等全身症状为主。虽然在西方常常有神经和皮肤受累[2],但在东亚相对少见,因此与SD鉴别相当困难。
根据日本的一项研究:相对IVL,SD患者主要是铁蛋白、IL-18有显著增高,而IVL患者有显著的sIL-2R增高,尽管SD患者的sIL-2R也有增高,但幅度远不如IVL[21],但最终的鉴别仍依赖于病理诊断。
7
感染性疾病、肿瘤性疾病
结核感染、肾细胞癌、淋巴瘤、白血病等等都可能带来长程发热。根据先定位(找病灶),再定性(定疾病诊断)的方式可以帮助医生更好的明确诊断。
总的来说,对各类长程发热疾病越熟悉,越不容易发生误诊。
治 疗
SD患者治疗应尽量实现疾病临床非活动性(CID),CID的定义是指任何时间点[1]:
没有任何SD相关表现,包括正常急性期反应物,如血沉(ESR)或CRP。如果进行多次急性期反应物测量,则所有措施都必须正常(除非存在其他原因来解释异常值,例如因为贫血而引发的ESR稍高)。
而临床缓解可定义为:CID维持≥6个月,无论治疗状态如何。
治疗的最终目标是无药物治疗下的临床缓解。而为实现这一目标,2024年EULAR/PReS联合指南建议设置的中间目标[1]:
- 第7天,发烧消退,CRP降低>50%;
- 第4周时,无发烧,活动(或肿胀)关节计数减少>50%,CRP正常,医生和患者/家长对视觉模拟评分法(VAS,0–100)的总体评估低于20;
- 在第3个月,糖皮质激素剂量低于0.1(成人)或0.2(儿童)mg/kg/天的CID;
- 在第6个月,没有糖皮质激素应用的CID。
根据目前的研究,对于任何确诊SD的患者,应尽快启动IL-1或IL-6抑制剂[1]。
在确诊前,阿司匹林以外的非甾体抗炎药(NSAID)均可用于控制症状。尝试NSAID单药治疗一般不应超过1-2周,因为在此期间通常很快即可看出是否需要加用其他药物[1-2]。
在最初发病时,IL-1或IL-6抑制剂与糖皮质激素联合使用是常见的。但要尽快减量糖皮质激素,从而控制糖皮质激素的副反应。
在使用IL-1或IL-6抑制剂的时代,甲氨蝶呤的使用在逐步减少。目前仅限于协助IL-1或IL-6抑制剂治疗,从而减少糖皮质激素的使用。在减量IL-1或IL-6抑制剂前,应确保无激素CID维持3-6个月[1]。
实际上,与IL-1抑制相比,IL-6抑制期间感染性不良事件(重症和非重症)更常见[1]。
就如前述,IL-18是重要的病理生理意义,也是诊断的重要标志物。那么靶向IL-18的治疗就成为很自然的选择。但很可惜的是,抑制IL-18的重组结合蛋白治疗成人发病的斯蒂尔病(AOSD:NCT02398435 II期)于2016年7月完成,初步证实了其安全性[23],但后续没有新的动向。
AVTX 007是一种抗IL-18单克隆抗体,专为治疗自身炎症性疾病而开发。早期临床开发正在进行中(NCT04752371),期待后续结果[24]。
APB R3是一种长效重组融合蛋白,由IL-18BP与抗人血清白蛋白Fab片段融合。关于其在治疗AOSD中的作用的研究正在进行中[24]。
Rood等描述了一份病例报告,其中IL-1β/IL-18联合阻断是SJIA-LD(难治性全身性幼年特发性关节炎相关肺病)的一种新治疗方法,初步看来是安全有效的[24]。但还需要更多研究来明确其临床价值。
Emapalumab是一种抗IFN-γ抗体,已经在SD相关的MAS治疗试验里得到初步验证[25-26],并被FDA批准。Emapalumab能否用于未并发MAS的单纯SD?尚需更多研究证据提供支撑。
至于MAS和SD-LD治疗,在后续篇章详细讨论。
往期回顾:
参考文献(上下滑动查看):
[1] Fautrel B, Mitrovic S, De Matteis A, et al. EULAR/PReS recommendations for the diagnosis and management of Still's disease, comprising systemic juvenile idiopathic arthritis and adult-onset Still's disease . Ann Rheum Dis. 2024 Nov 14;83(12):1614-1627.
[2] Uptodate临床顾问
[3] Chen DY, Lan HH, Hsieh TY, et al. Crico-thyroid perichondritis leading to sore throat in patients with active adult-onset Still's disease. Ann Rheum Dis 2007; 66:1264.
[4] Arianna De Matteis , Bruno Fautrel , Stéphane Mitrovic , et al. Systemic juvenile idiopathic arthritis and adult-onset Still’s disease are the same disease: evidence from systematic reviews and meta-analyses informing the 2023 EULAR/PReS recommendations for the diagnosis and management of Still’s disease .Ann Rheum Dis. 2024 Sep 24;83(12):e225853.
[5] Verena Schoenau ,et al. Patients with Adult-Onset Still’s Disease in Germany: A Retrospective Analysis of Clinical Characteristics and Treatment Practices Ahead of the Release of the German Recommendations . J Clin Med. 2025 Feb 4;14(3):981.
[6] Efthimiou P, Kontzias A, Hur P, et al. Adult-onset Still's disease in focus: Clinical manifestations, diagnosis, treatment, and unmet needs in the era of targeted therapies. Semin Arthritis Rheum 2021; 51:858.
[7] Lee PY, Schulert GS, Canna SW, et al. Adenosine deaminase 2 as a biomarker of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis 2020; 79:225.
[8] Lee PY, Davidson BA, Abraham RS, Alter B, Arostegui JI, Bell K, et al. Evaluation and Management of Deficiency of Adenosine Deaminase 2: An International Consensus Statement. JAMA Netw Open. 2023;6(5): e2315894.
[9] Yin J, Fan X, Ma J, Liu X, Li C. ADA2 deficiency (DADA2) misdiagnosed as systemic onset juvenile idiopathic arthritis in a child carrying a novel compound heterozygousADA2mutation: a case report . Transl Pediatr. 2023 Jan 31;12(1):97-103.
[10] Girard-Guyonvarc'h C, Rodriguez E, Mueller YM, Caruso A, Katsikis PD, Gabay C; ImmunAID consortium . Elevated serum levels of interleukin-18 discriminate Still's disease from other autoinflammatory conditions: results from the European ImmunAID cohort.RMD Open. 2025 May 7;11(2):e005388.
[11] Firestein Kelley’s Textbook of Rheumatology(第12版)
[12] Wakil, S.M., Monies D.M., Al-Mayouf S,et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 67:288–295.
[13] Cader, M.Z., Boroviak K., Zhang Q., Assadi G., Kempster S.L., Sewell G.W., Saveljeva S., Ashcroft J.W., Clare S., Mukhopadhyay S., et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat. Immunol. 17:1046–1056.
[14] Skon-Hegg, C., Zhang J., Wu X., Caplazi P., et al. LACC1 Regulates TNF and IL-17 in Mouse Models of Arthritis and Inflammation. J. Immunol. 202:183–193.
[15] Sulaiman M Al-Mayouf , Mada Yateem , Haya Al-Dusery , et al. New or vanishing frontiers:LACC1-associated juvenile arthritis . Int J Pediatr Adolesc Med. 2021 Mar;8(1):44-47.
[16] Tilmann Kallinich , Anne Thorwarth , Sae-Lim von Stuckrad , et al. Juvenile arthritis caused by a novelFAMIN (LACC1)mutation in two children with systemic and extended oligoarticular course . Pediatr Rheumatol Online J. 2016 Nov 24;14:63.
[17] Yali Wu , Shasha Wang , Wen Yin , Wei Yin , Yan Ding . Clinical characteristics and genotype analysis of a Chinese patient with juvenile arthritis due to novel LACC1 frameshift mutation and literature review . Mol Genet Genomic Med. 2023 Jul;11(7):e2175.
[18] Tingyan He ,Linlin Wang ,Xiaomei Huang,Ruohang Weng,Jun Yang . LACC1 deficiency leading to juvenile arthritis and anemia . Clin Immunol. 2024 Aug:265:110290.
[19] Ommar Omarjee , Anne-Laure Mathieu , Gaëlle Quiniou , et al. LACC1 deficiency links juvenile arthritis with autophagy and metabolism in macrophages . J Exp Med. 2021 Mar 1;218(3):e20201006.
[20] Oliveira JB, Bleesing JJ, Dianzani U, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 2010; 116:e35.
[21] Takahiro Shima , Yusuke Yamauchi, Masahiro Ohtsu , et al. Evaluation of clinical biomarkers for differential diagnosis of intravascular lymphoma and Still’s disease . Sci Rep. 2025 Jul 4;15:23901.
[22] Bindoli S, De Matteis A, Mitrovic S, et al. Efficacy and safety of therapies for the treatment of adult-onset still’s disease (AOSD) and systemic juvenile idiopathic arthritis (sJIA): a systematic review informing the EULAR PRES guidelines for the diagnosis and management of systemic juvenile idiopathic arthritis and adult-onset still’s disease. Rev. 2024
[23] Gabay C, Fautrel B, Rech J, et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still's disease. Ann Rheum Dis 2018; 77:840.
[24] Baggio C, Bindoli S, Guidea I, Doria A, Oliviero F, Sfriso P. IL-18 in Autoinflammatory Diseases: Focus on Adult Onset Still Disease and Macrophages Activation Syndrome.Int J Mol Sci. 2023 Jul 5;24(13):11125.
[25] De Benedetti F, Brogan P, Grom A, et al. Empalumab, an interferon gamma (IFN-g)-blocking monoclonal antibody, in patients wtih macrophage activation syndrome (MAS) complicating systemic juvenile idiopathic arthritis (sJIA). OP0204. Ann Rheum Dis 2018; 78:178.
[26] De Benedetti F, Grom AA, Brogan PA, et al. Efficacy and safety of Emapalumab in macrophage activation syndrome. Ann Rheum Dis. 2023;82:857–65.


